Atmospheric nitrate (particulate nitrate + gaseous HNO3) plays an essential role in the biogeochemical cycle of nitrogen in the Arctic region. However, there are poor studies on the formation mechanisms of atmospheric nitrate over the Arctic Ocean, which limits our understanding of atmospheric chemical processes related to nitrogen oxides (NOX=NO+NO2) and nitrate in this area. As part of 5th Chinese National Arctic Research Expedition (CHINARE) in the summer of 2012, aerosol filter samples were collected and nitrogen and oxygen isotopes of nitrate (δ15N, δ17O and δ18O) in the filter samples in 62.3°~74.7° N were analyzed to trace the formation pathways of atmospheric nitrate over the Arctic Ocean. The observed daily Δ17O(NO) (=δ17O–0.52δ18O) varied from 21.7‰ to 28.8‰ with the mean of (25.4±2.7)‰ and δ15N(NO) ranged from -7.5‰ to 0.8‰ with the mean of (-4.2±3.0)‰. Generally speaking, Δ17O(NO) showed a opposite trend with the sampling latitude, and a similar trend with the nighttime hours and O3 concentration. While δ15N(NO) showed the opposite trend with air temperature. Chemical kinetics calculations show that the variation of Δ17O(NO) may be mainly determined by the role of different pathways in nitrate production rather than the relative importance of O3 and XO (X=Br, Cl and I) in NOX cycling, the latter was estimated to be 0.81~0.90 with the mean of 0.85±0.03. Further analysis based on Δ17O(NO) showed that NO2+OH pathway dominated nitrate production for samples with low Δ17O(NO) (=21.7‰~24.5‰, 66.2°~74.7° N), with the mean possible contribution of 68%~81%. For samples with relatively high Δ17O(NO) (=27.5‰~28.8‰, 62.3°~69.9° N), the together role of NO3+HC/DMS, NO3+H2O(aq) and XNO3+H2O(aq) are the most important, with the mean possible contribution of 35%~50%. Combined with the analysis of BrO column concentrations, it was found that the role of XNO3+H2O(aq) in nitrate production cannot be ignored for high Δ17O(NO) samples (e.g., Δ17O=28.8‰), the role of which needs to be further explored with the combination of atmospheric chemistry model in future studies.
HE Pengzhen, XIE Zhouqing. Using oxygen isotopes to trace the formation pathways of atmospheric nitrate over summer Arctic Ocean (62.3°~74.7° N)[J]. Journal of Glaciology and Geocryology, 2021, 43(5): 1344-1353 doi:10.7522/j.issn.1000-0240.2021.0088
Fig.1
The diagram of nitrate formation pathways in global scale (Numbers in the brackets show the global annual-mean contribution to NO2 and nitrate formation from model[7]. X represents Cl, Br and I, HC and DMS represents hydrocarbons and dimethyl sulfide, respectively. MTN and ISOP represents monoterpenes and isoprene, respectively. The grey and yellow areas represent nitrate formation pathways that only significant in nighttime and in daytime, respectively)
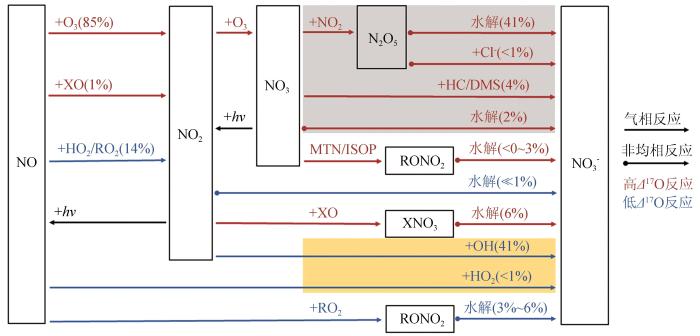
Fig.2
The observation of atmospheric Δ17O(NO) and δ15N(NO) along the cruise over the Arctic Ocean during 5th Chinese National Arctic research Expedition in summer 2012; The gray level in the background represents the ocean bathymetry[42]
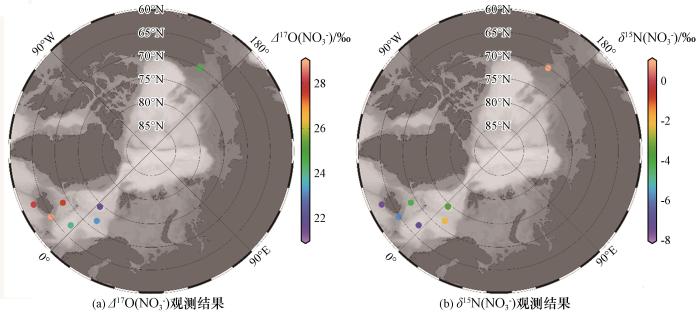
Fig.3
The variation of isotopes with latitudes (a), solar radiation (b), ozone mixing ratio (c) and atmospheric temperature (d) during our observations
Fig. 4
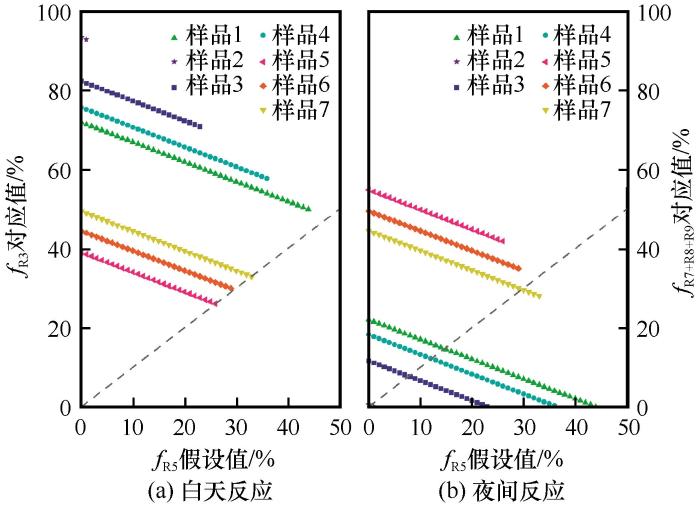
The estimate of possible fractional contribution of different formation pathways to nitrate production based on Δ17O(NO), R3, R5, R7~R9 represents NO2+OH, N2O5+H2O(aq), NO3+HC/DMS, NO3+H2O(aq) and XNO3+H2O(aq), respectively. The dash line is for 1:1
Fig. 5
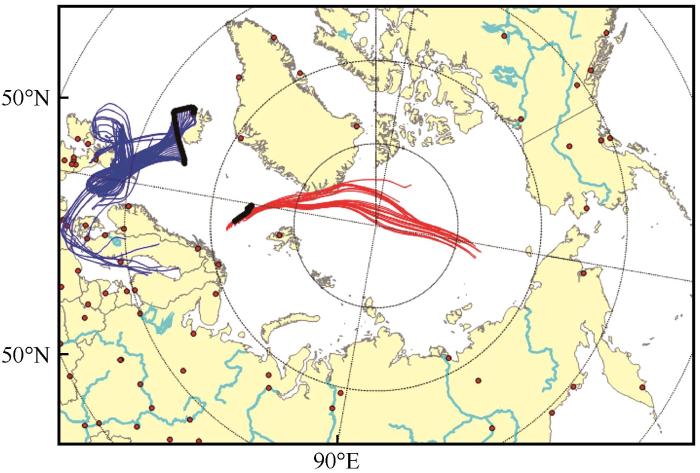
The backward trajectory analysis of air mass during the collection of featured aerosol samples. The red lines represent backward trajectories of air mass during the collection of Sample 2 in August 6—7 when Δ17O(NO) is the lowest while the blue lines represent backward trajectories of air mass during the collection of Samples 5~6 in August 12—14 when Δ17O(NO) are the highest. The running time and altitude for each of the backward trajectory analysis is 5 days and 50 m, respectively. The black dot-lines represent locations of the sampling covered, the red dots are cities on land. This figure was drawn by TrajStat[44], in which the model of HYSPLIT and meteorological data of 1°×1° from NOAA was used
Fig.6
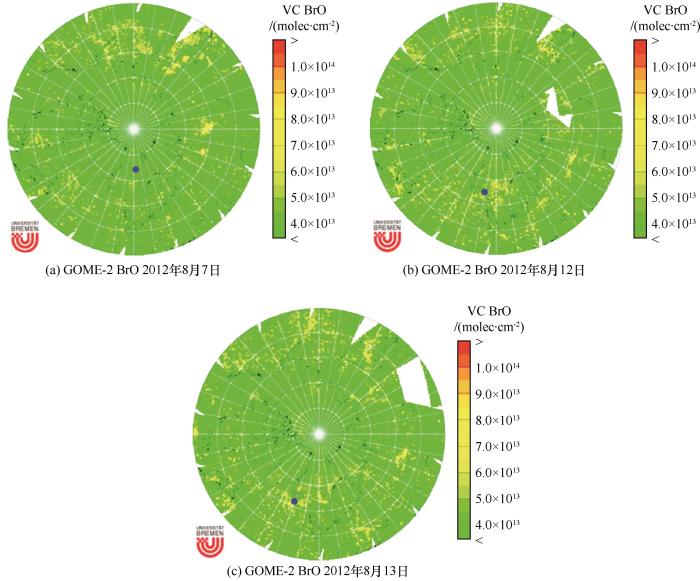
The vertical column concentration of BrO observed by satellite. The blue dot represents the median location of the filter sampling. The BrO figures were modified fromhttp://www.iup.physik.uni-bremende/doas/scia_data_browser.htm(2021-07-06 accessed)
A factor and trends analysis of multidecadal lower tropospheric observations of arctic aerosol composition, black carbon, ozone, and mercury at alert, canada
[J]. Journal of Geophysical Research-Atmospheres, 2019, 124(24): 14133-14161.
Changes in nitrate accumulation mechanisms as PM2.5 levels increase on the north china plain: A perspective from the dual isotopic compositions of nitrate
Quantitative constraints on the 17O-excess (Δ17O) signature of surface ozone: ambient measurements from 50° N to 50° S using the nitrite-coated filter technique
The NO+O3 reaction: a triple oxygen isotope perspective on the reaction dynamics and atmospheric implications for the transfer of the ozone isotope anomaly
[J]. Journal of Chemical Physics, 2008, 128(19): 194303.
Investigation of post-depositional processing of nitrate in east antarctic snow: Isotopic constraints on photolytic loss, re-oxidation, and source inputs
[J]. Atmospheric Chemistry and Physics, 2015, 15(16): 9435-9453.
TrajStat: GIS-based software that uses various trajectory statistical analysis methods to identify potential sources from long-term air pollution measurement data
... [7].X代表元素Br、Cl和I,HC和DMS代表碳氢化合物和二甲基硫,MTN和ISOP分别代表单萜和异戊二烯.灰色阴影区域代表只发生在夜间硝酸盐的形成途径,黄色阴影区域代表只发生在白天的硝酸盐的形成途径)The diagram of nitrate formation pathways in global scale (Numbers in the brackets show the global annual-mean contribution to NO<sub>2</sub> and nitrate formation from model<sup>[<xref ref-type="bibr" rid="R7">7</xref>]</sup>. X represents Cl, Br and I, HC and DMS represents hydrocarbons and dimethyl sulfide, respectively. MTN and ISOP represents monoterpenes and isoprene, respectively. The grey and yellow areas represent nitrate formation pathways that only significant in nighttime and in daytime, respectively)Fig.1
... [7]. X represents Cl, Br and I, HC and DMS represents hydrocarbons and dimethyl sulfide, respectively. MTN and ISOP represents monoterpenes and isoprene, respectively. The grey and yellow areas represent nitrate formation pathways that only significant in nighttime and in daytime, respectively)Fig.1
A factor and trends analysis of multidecadal lower tropospheric observations of arctic aerosol composition, black carbon, ozone, and mercury at alert, canada
Changes in nitrate accumulation mechanisms as PM2.5 levels increase on the north china plain: A perspective from the dual isotopic compositions of nitrate
Quantitative constraints on the 17O-excess (Δ17O) signature of surface ozone: ambient measurements from 50° N to 50° S using the nitrite-coated filter technique
The NO+O3 reaction: a triple oxygen isotope perspective on the reaction dynamics and atmospheric implications for the transfer of the ozone isotope anomaly
Investigation of post-depositional processing of nitrate in east antarctic snow: Isotopic constraints on photolytic loss, re-oxidation, and source inputs
The observation of atmospheric <i>Δ</i><sup>17</sup>O(NO<span class="formulaText"><inline-formula><math id="M56"><msubsup><mrow/><mrow><mn mathvariant="normal">3</mn></mrow><mrow><mo>–</mo></mrow></msubsup></math></span></inline-formula></span>) and <i>δ</i><sup>15</sup>N(NO<span class="formulaText"><inline-formula><math id="M57"><msubsup><mrow/><mrow><mn mathvariant="normal">3</mn></mrow><mrow><mo>–</mo></mrow></msubsup></math></span></inline-formula></span>) along the cruise over the Arctic Ocean during 5th Chinese National Arctic research Expedition in summer 2012; The gray level in the background represents the ocean bathymetry<sup>[<xref ref-type="bibr" rid="R42">42</xref>]</sup>Fig.2
观测期间硝酸盐氮氧同位素数据与纬度(a),太阳辐射(b),臭氧(c)和气温(d)的变化趋势
The variation of isotopes with latitudes (a), solar radiation (b), ozone mixing ratio (c) and atmospheric temperature (d) during our observationsFig.3
TrajStat: GIS-based software that uses various trajectory statistical analysis methods to identify potential sources from long-term air pollution measurement data
The estimate of possible fractional contribution of different formation pathways to nitrate production based on <i>Δ</i><sup>17</sup>O(NO<span class="formulaText"><inline-formula><math id="M77"><msubsup><mrow/><mrow><mn mathvariant="normal">3</mn></mrow><mrow><mo>–</mo></mrow></msubsup></math></span></inline-formula></span>), R3, R5, R7~R9 represents NO<sub>2</sub>+OH, N<sub>2</sub>O<sub>5</sub>+H<sub>2</sub>O(aq), NO<sub>3</sub>+HC/DMS, NO<sub>3</sub>+H<sub>2</sub>O(aq) and XNO<sub>3</sub>+H<sub>2</sub>O(aq), respectively. The dash line is for 1:1Fig. 4
The backward trajectory analysis of air mass during the collection of featured aerosol samples. The red lines represent backward trajectories of air mass during the collection of Sample 2 in August 6—7 when <i>Δ</i><sup>17</sup>O(NO<span class="formulaText"><inline-formula><math id="M80"><msubsup><mrow/><mrow><mn mathvariant="normal">3</mn></mrow><mrow><mo>–</mo></mrow></msubsup></math></span></inline-formula></span>) is the lowest while the blue lines represent backward trajectories of air mass during the collection of Samples 5~6 in August 12—14 when <i>Δ</i><sup>17</sup>O(NO<span class="formulaText"><inline-formula><math id="M81"><msubsup><mrow/><mrow><mn mathvariant="normal">3</mn></mrow><mrow><mo>–</mo></mrow></msubsup></math></span></inline-formula></span>) are the highest. The running time and altitude for each of the backward trajectory analysis is 5 days and 50 m, respectively. The black dot-lines represent locations of the sampling covered, the red dots are cities on land. This figure was drawn by TrajStat<sup>[<xref ref-type="bibr" rid="R44">44</xref>]</sup>, in which the model of HYSPLIT and meteorological data of 1°×1° from NOAA was usedFig. 5
The vertical column concentration of BrO observed by satellite. The blue dot represents the median location of the filter sampling. The BrO figures were modified from<ext-link ext-link-type="uri" xlink:href="http://www.iup.physik.uni-bremen">http://www.iup.physik.uni-bremen</ext-link>de/doas/scia_data_browser.htm(2021-07-06 accessed)Fig.6
The vertical column concentration of BrO observed by satellite. The blue dot represents the median location of the filter sampling. The BrO figures were modified from<ext-link ext-link-type="uri" xlink:href="http://www.iup.physik.uni-bremen">http://www.iup.physik.uni-bremen</ext-link>de/doas/scia_data_browser.htm(2021-07-06 accessed)Fig.6


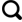



 甘公网安备 62010202000676号
甘公网安备 62010202000676号
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}